Static headspace enantioselective comprehensive two-dimensional gas chromatography–time-of-flight mass spectrometry in food analysis: a proof-of-principle study
Analytica Chimica Acta, 1401, 2026, 345335: Graphical abstract
This study evaluates static headspace sampling combined with enantioselective comprehensive two-dimensional GC–TOFMS for volatile analysis in food matrices. Using bergamot and sweet orange essential oils as model samples, the method enabled solvent-free profiling of volatile compounds and determination of enantiomeric ratios.
Although static headspace showed reduced detectability compared to direct injection, cryogenic modulation partly compensated for sensitivity loss. Comparable chiral profiles were obtained with both approaches, demonstrating the potential of SHS eGC×GC–TOFMS for authenticity assessment, origin determination, and transformation product analysis in food applications.
The original article
Static headspace enantioselective comprehensive two-dimensional gas chromatography–time-of-flight mass spectrometry in food analysis: a proof-of-principle study
Micaela Galletta, Mariosimone Zoccali, Ivana L. Bonaccorsi, Peter Q. Tranchida, Luigi Mondello
Analytica Chimica Acta, 1401, 2026, 345335
https://doi.org/10.1016/j.aca.2026.345335
licensed under CC-BY 4.0
Selected sections from the article follow. Formats and hyperlinks were adapted from the original.
Static headspace gas chromatography-mass spectrometry (SHS GC-MS) is a convenient means to analyze the volatile fraction of food products (and not only) because: I) it can provide a reliable view on the volatile compounds that leave the surface of a food and reach the human nose, eliciting a biological response (aroma); II) volatile products of transformation (e.g., due to oxidation) can be potentially detected, providing a view on the state of preservation; III) it is a simple sampling technique free of any sample preparation step (hence organic solvents are not required) [[1], [2], [3]]. During SHS sampling, equilibrium between the sample and the headspace above it is achieved, and a fraction of the headspace gas phase is withdrawn for GC analysis [4]. Although SHS is often characterized by lower analyte detectability compared to enrichment-based techniques [5], such as solid-phase microextraction (SPME), stir-bar sorptive extraction (SBSE) and dynamic headspace (DHS) sampling [6], it represents an equilibrium-based sampling approach that does not involve an absorption or adsorption step, thereby minimizing analyte discrimination effects. In contrast, SPME and SBSE discriminate between analytes with different affinities for the absorption/adsorption stationary phase [7,8]. Furthermore, while DHS enables exhaustive sampling, it operates under non-equilibrium conditions by continuously purging the vial headspace onto a sorbent-containing trap. Once the trap is thermally-desorbed, the released analytes may, or may not be, focused by using a cooled inlet. Artifact formation may arise during DHS sampling, especially for samples containing water [9]. Multiple SHS sampling processes can also be performed to increase analyte detectability; also in this case a trapping process is necessary [10].
Within the context of SHS GC-MS, Deng et al. performed the analysis of the leaves of 42 Citrus cultivars. The SHS incubation time and temperature were 15 min and 100 °C [11]; considering all the cultivars, a total of 83 compounds were identified. The use of an above-ambient temperature increased analyte concentrations in the headspace, also for compounds with lower vapour pressures. For such a reason, the obtained results did not reflect the ambient-temperature HS, which is an important aspect in food analysis. In fact, high temperatures will lead to the detection also of semi-volatile compounds and accelerate processes of transformation.
The use of SHS sampling, combined with cryogenic modulation comprehensive two-dimensional gas chromatography (CM GC × GC), has been rarely reported. For example, Heng et al. used SHS CM GC × GC to monitor the presence of Escherichia coli in processed dairy milk [12]. The SHS CM GC × GC approach is interesting, because cryogenic-focusing between the first (1D) and second (2D) dimension increases signal-to-noise ratio (s/n) values, thus analyte detectabilities. The enhancement of s/n values is variable, as it depends on several factors such as modulation ratio, dimension of the 2D column, 2D column temperature and gas flow, and 2D analye-stationary phase affinity [13].
The authenticity of essential oils represents a critical issue in quality control, largely due to the diffusion of adulteration driven by the high global demand for these products [14]. In such a respect, the continuous expansion of the global market increases the need of advanced analytical methodologies to assess authenticity. Enantioselective gas chromatography (eGC) is a well-established and widely-used technique for the characterization of essential oils [15,16]. In particular, specific enantiomeric ratios are reported for key components of Citrus essential oils [15]. The column combination herein used, enabled the attainment of both a general sample fingerprint and the separation of specific chiral compounds, in a single analysis [17]. For such a reason, eGC × GC can be considered as a superior alternative to heart-cutting enantioselective multidimensional GC.
The present study evaluates the applicability of SHS sampling prior to CM eGC × GC combined with time-of-flight MS (ToFMS) for the simultaneous untargeted and chiral profiling of Citrus essential oils. The analytical approach used can be extended to other types of food samples for which enantioselective analysis is important [18].
2. Materials and methods
2.2. Instrumentation
The SHS and DI processes were performed automatically by using an L-PAL3 GC Autosampler (LECO Corporation, St. Joseph, MI, USA). For DI, the injection volume was 1 μL using a split ratio of 1:10 at 220 °C. In all SHS applications, 0.5 mL of essential oil (previously brought to a room temperature of 26 °C) was transferred to a 10 mL vial, which was then sealed. The samples were incubated for 10 min at 30 °C with an agitation speed of 300 rpm. The optimized SHS parameters were as follows: 30 s pre-purge syringe time, 80 °C syringe temperature, 20 mL min−1 injection flow rate, 0.5 s post-injection dwell time, and an injection volume of 1 mL. Three replicates for each sample were carried out. The eGC × GC-ToFMS applications were performed on a Pegasus® BT 4D GC × GC-ToFMS system equipped with a QuadJet™ thermal modulator (LECO Corporation). The GC × GC column set was: Merck Astec® CHIRALDEX™ B-DM (catalog n. 77023AST) [2,3-di-O-methyl-6-t-butyl silyl derivative of β-cyclodextrin phase] with dimensions 30 m × 0.25 mm ID × 0.12 μm df used as 1D column, while the 2D column was a segment of an SLB-5ms (catalog n. 28564-U) [silphenylene polymer with similar polarity to poly (5% diphenyl/95% dimethylsiloxane)] with dimension 1.75 m × 0.18 mm ID × 0.18 μm df, and with 0.3 m located inside the MS transfer line. All columns were provided by Merck Life Science. Helium was used as a carrier gas and was delivered at a constant flow of 1.2 mL min−1. The main oven temperature program was 50 °C, then ramped up to 220 °C at 1 °C min−1. The temperature offset for the secondary oven and the modulator were set at +25 °C and +15 °C, with respect to the main oven temperature, respectively. A modulation period of 3 s was used, with 0.90 s hot jet and 0.60 s cold jet times. The transfer line and the ion source temperatures were set at 220 and 250 °C, respectively. A mass range from 40 to 400 m/z was applied with an acquisition rate of 150 spectra s−1. The detector voltage was that relative to the tuning process; solvent delay was 260 s. Electron ionization was performed at 70 eV. Data were collected and processed using the ChromaTOF® BT software version 5.56.42 (LECO). The mass spectral database used was the Flavour and Fragrance Natural and Synthetic Compounds FFNSC 4 (Shimadzu Europa, Duisburg, Germany).
3. Results and discussion
3.1. Untargeted analysis
The developed method enabled an in-depth investigation of the volatile composition of the two cold-pressed essential oil samples. The total-ion-current (TIC) chromatograms of the DI and SHS CM eGC × GC-ToFMS analysis of bergamot essential oil are shown in Fig. 1A and B. The differences between the chromatographic profiles are readily visible, and relate to the aspects previously discussed. The chromatograms have been divided into different temperature zones of elution to make a visual comparison easier. As to be expected, the SHS chromatogram clearly presents a lower number of detected compounds.
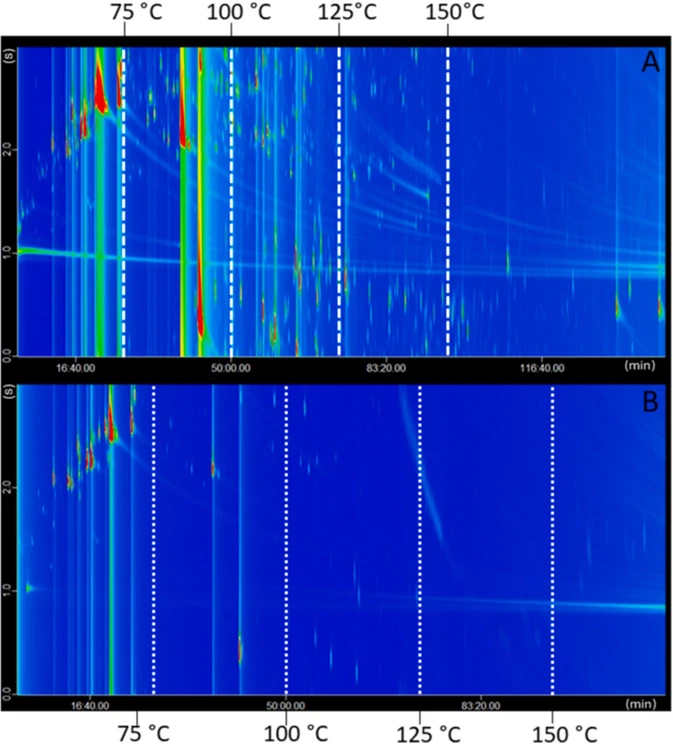 Analytica Chimica Acta, 1401, 2026, 345335: Fig. 1. The TIC chromatograms relative to the DI (A) and SHS (B) CM eGC × GC-ToFMS analysis of bergamot essential oil. Elution temperature zones are also indicated.
Analytica Chimica Acta, 1401, 2026, 345335: Fig. 1. The TIC chromatograms relative to the DI (A) and SHS (B) CM eGC × GC-ToFMS analysis of bergamot essential oil. Elution temperature zones are also indicated.
The bergamot oil volatile fraction (approximately 95% of the whole oil) is represented by monoterpene and sesquiterpene hydrocarbons, and their oxygenated derivatives, along with aliphatic aldehydes, alcohols, and esters [19].
Initially, attention was devoted to untargeted analysis (considering a minimum spectral forward MS similarity ≥800). Overall, a total number of 239 and 84 analytes were tentatively-identified using DI and SHS, respectively (Table S1). The identity of a series of analytes was confirmed by injecting the corresponding pure reference standard compound (section 2.1).
The last-eluting tentatively-identified compound using DI was bergapten (C12H8O4 - 216.19 g mol−1, with an elution temperature of about 191 °C), while for SHS it was β-bisabolene (C15H24 - 204.35 g mol−1, with an elution temperature of about 122 °C).The SHS technique is based on Henry's law, inasmuch that the concentration of a specific volatile in the vial HS depends on its sample-HS distribution coefficient, sample concentration, vapour pressure, along with the volumes of the HS and sample [9]. Boiling points (along with molecular weights) will be herein used as an expression of vapour pressure, hence analyte volatility.
The two most abundant bergamot essential oil compounds, in both cases, were linalyl acetate (C12H20O2 - 196.29 g mol−1) and limonene: 2 (C10H16 -136.23 g mol−1). However, when using DI and SHS the peak area % values for linalyl acetate were 30.43 and 10.12%, respectively, and for limonene: 2, 18.21% and 29.59%, respectively. Such an inversion relates to the higher volatility of limonene [boiling point (b.p.): 176 °C at 1 atm), in contrast to linalyl acetate (b.p.: 220 °C at 1 atm), despite the latter being the major constituent of the essential oil.
A further comparison between the two analytical approaches revealed that the % peak area of linalol: 1 (C10H18O) decreased from 7.98% to 1.89% when using SHS, whereas γ-terpinene (C10H16) increased slightly from 5.50% to 6.36%. Additionally, α-pinene and β-pinene (C10H16) % values were higher when using the SHS procedure, in contrast to p-cymene (C10H14), which diminished slightly. All other constituents were present at levels below 1%. In bergamot essential oil, both linalyl acetate and linalol are present at relatively high levels compared to other Citrus oils [19]. The organoleptic characteristics and genuineness of bergamot essential oil are usually defined by the linalol/linalyl acetate ratio and the sum of these two constituents [20,21].
Bergapten (C12H8O4) and citropten (C11H10O4), oxygen heterocyclic compounds, were as expected not detected in the SHS analysis (while they were in the DI one). Such constituents are recognized as key quality markers of Citrus oils, with their determination also important due to their potential toxicity to humans [22]. It is noteworthy that the analysis of oxygen heterocyclic compounds is normally carried out by using high-performance liquid chromatography [23].
With regard to sesquiterpenes, only four (trans-α-bergamotene, β-caryophyllene, α-humulene, and β-bisabolene) were detected when using SHS, whereas 52 were identified using DI.
The number of tentatively-identified compounds using DI CM eGC × GC was obviously much higher compared to straightforward DI GC-MS, and for such a reason such information can be used to determine genuineness (or not), geographical origin, or the presence of products of oxidation. Conversely, the number of tentatively-identified compounds using SHS CM eGC × GC was comparable to DI GC-MS (usually between 80 and 100 compounds) [24], and can be exploited for the same scopes.
A series of components of Citrus essential oils are well known to be susceptible to oxidative deterioration during storage. For instance, linalol and limonene can autoxidize to form hydroperoxides, which are recognized as potential allergens [25]. In the present study, the cis-limonene oxide enantiomers (C10H16O) were detected only in the DI analysis, whereas those of cis-linalol oxide (C10H18O2) were identified using both sampling approaches; for each compound, MS similarity values ≥ 900 were obtained (Table S1). Furthermore, the formation of geranyl and neryl acetates (C12H20O) in bergamot oil can be attributed to an intramolecular rearrangement reaction of linalyl acetate [26]. Both compounds (trans isomer for geranyl acetate) were found when using DI and SHS. Likewise, the occurrence of (E)-β-ocimene (C10H16) can be explained by the thermo-oxidative degradation pathway of linalyl acetate, involving acetic acid elimination followed by rearrangement of the monoterpene [26]. This compound was detected exclusively in the SHS analysis with a similarity value of 885, whereas it was not detected in the DI analysis due to coelution with the second enantiomer of limonene. The peak area of limonene: 2 was substantially greater in the DI analysis. It is presumable that the detected oxidation products were probably already present in the sample, resulting from natural aging and exposure to environmental factors, rather than being formed during the analytical procedure.
4. Conclusions
The present research indicates that SHS sampling, prior to CM eGC × GC-ToFMS, is a method of interest for the untargeted and chiral characterization of Citrus essential oils. Only rare studies have been focused on the SHS GC analysis of Citrus essential oils, while DI GC has been by far the predominant technique used. For such a reason, especially for untargeted analysis, a direct comparison for the scope of authenticity confirmation, cannot be made (the literature contains basically only DI data).
It is the present Authors’ opinion that the option SHS CM eGC × GC-MS could potentially be of general interest in food analysis. However, such an applicability should be mainly related to volatile-rich and chemically-complex matrices, in which aroma-impact compounds play a central role.
Apart from the “cleaness” of SHS sampling, it can provide a good view on the aroma-impact volatiles that reach the human nose; further, the untargeted information can be used to confirm authenticity and define geographical origin (if a wide range of SHS data becomes available), along with the determination of products of transformation. Finally, the information on enantiomer distribution can be exploited for authenticity investigations, consulting data already present in the literature. Conversely, SHS discriminates between more and less-volatile compounds and could present limitations when applied to samples characterized by low concentrations of volatiles and/or with a dominating presence of constituents with higher b.p. values.
For SHS studies, involving contaminants (e.g., pesticide, phthalates, etc.), the sample would need to be heated. With regard to analyte quantification, this can be carried out, among others, through standard addition [9].
Future research will be devoted to the extension of use of SHS CM eGC × GC-MS in food analysis.